
Traduction d’après un article de Spectrum
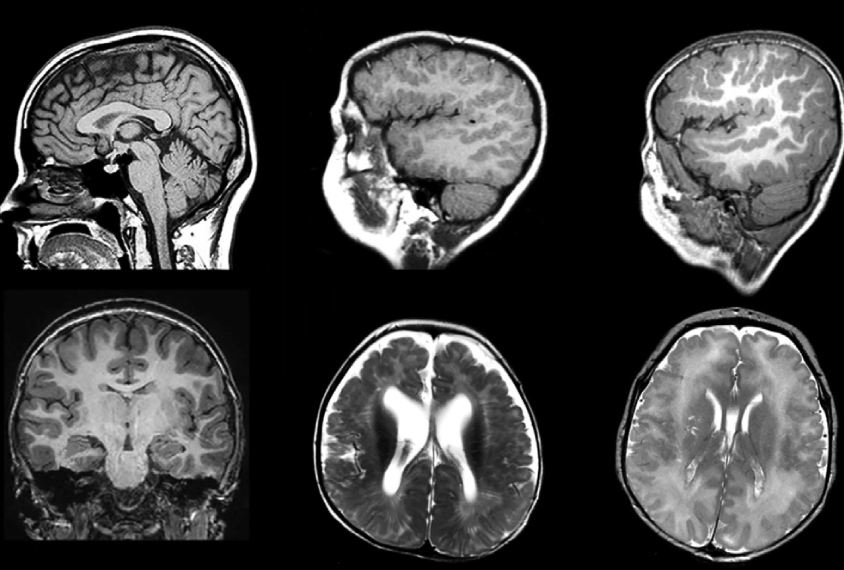
Les enfants présentant des mutations dans un gène appelé DDX3X ont des plis cérébraux inhabituellement petits.
Les chercheurs ont recensé plus de 100 mutations du gène DDX3X, un gène candidat pour l’autisme (1). Cette étude est la première à analyser de près la relation entre ces mutations et leurs effets sur les personnes autistes.
Les chercheurs ont combiné des observations cliniques et des scanners cérébraux d’enfants avec des analyses génétiques chez la souris afin de déterminer la base moléculaire des traits des enfants.
Les chercheurs ont établi un premier lien entre le DDX3X et l’autisme en 2014, et avec la déficience intellectuelle l’année suivante (2,3). Le gène étant situé sur le chromosome X, les mutations touchent principalement les filles ; on pense que la plupart des garçons présentant des mutations dans le gène meurent in utero.
Jusqu’à 3 % des filles présentant une déficience intellectuelle inexpliquée présentent une mutation du gène DDX3X(3). Environ la moitié d’entre elles sont également susceptibles d’être atteintes d’autisme, selon le cochercheur principal Elliott Sherr, professeur de neurologie à l’université de Californie, à San Francisco.
« Les enfants qui présentent la forme la plus légère présenteraient un tableau clinique encore important », précise M. Sherr. Le gène DDX3X « est probablement très important pour réguler l’activation et la désactivation de gènes importants qui sont essentiels au développement précoce ».
L’étude a montré que différentes mutations dans le gène conduisent à des combinaisons variées et à la gravité de ces traits.
« C’est un élément clé à prendre en compte lorsque nous réfléchissons aux approches thérapeutiques du syndrome DDX3X », déclare Silvia De Rubeis, professeur adjoint de psychiatrie au Seaver Autism Center for Research and Treatment à New York, qui n’a pas participé aux nouveaux travaux.
Sherr et ses collègues ont analysé les génomes de 107 enfants présentant des mutations dans le gène DDX3X. Presque toutes les mutations sont apparues de novo, ou spontanément, plutôt que d’être héritées.
Environ un tiers sont des mutations de « non sens », ce qui signifie que le gène ne parvient pas à fabriquer la protéine correspondante. Et environ la moitié sont des mutations de « faux sens », qui conduisent à ce que le gène fabrique une protéine défectueuse. Les résultats ont été publiés le 4 mars dans Neuron.
Résultats génétiques :
La plupart des 57 enfants porteurs de mutations de « faux sens » présentent de graves caractéristiques, notamment une déficience intellectuelle, un faible tonus musculaire et une incapacité à marcher. Les scanners cérébraux montrent qu’ils présentent également de nombreux plis anormalement petits dans le cerveau, une condition connue sous le nom de polymicrogyrie.
Trois des enfants sont porteurs de la même mutation au même endroit de leur génome et présentent des traits similaires, ce qui montre que cette mutation est à l’origine des traits spécifiques, explique M. Sherr.
Vous pouvez toujours dire « Eh bien, c’est peut-être juste une coïncidence », mais le fait qu’il s’agisse exactement de la même mutation à plusieurs reprises et qu’ils aient tous le même phénotype est assez convaincant », déclare M. Sherr.
Les chercheurs ont ensuite utilisé la technique d’édition génétique CRISPR pour modifier le gène DDX3X chez des souris à différents stades de développement.
Les souris ayant de faibles niveaux d’expression du DDX3X ont beaucoup moins de neurones que les témoins, ce qui suggère que le gène est crucial pour générer des neurones au début du développement du cerveau, explique la co-chercheuse principale Debra Silver, professeur associé de génétique moléculaire et de microbiologie à l’université Duke de Durham, en Caroline du Nord.
« La conclusion de tous ces efforts est que le cerveau est extrêmement sensible aux niveaux de DDX3X », explique Mme Silver. « Même une réduction de 25 % des niveaux de DDX3X a un impact profond sur la production de neurones ».
Fonction neuronale :
Les chercheurs ont découvert que de faibles niveaux de DDX3X empêchent l’ARN de se dérouler correctement : Dans les neurones de souris et les cellules souches neurales, les mutations erronées provoquent l’accumulation de protéines.
Selon la Fondation DDX3X, une organisation à but non lucratif qui se consacre à cette maladie, moins de 500 personnes dans le monde sont atteintes du syndrome DDX3X. Beaucoup d’autres pourraient ne pas encore être identifiées, notamment parce que le lien entre ce gène et la déficience intellectuelle n’a été découvert qu’il y a environ cinq ans, explique M. Silver.
Au fur et à mesure que les chercheurs identifieront d’autres enfants porteurs de ces mutations, ils pourront peut-être mettre en évidence les effets de certaines d’entre elles.
Il sera important de comprendre ces relations pour s’assurer que les gens reçoivent les bons traitements pour les mutations particulières dont ils sont porteurs, explique Tychele Turner, professeur adjoint de génétique à l’université de Washington à St.
« Il est vraiment essentiel de s’intéresser à l’aspect fonctionnel de ces gènes, et pas seulement aux gènes mais aussi aux mutations qu’ils contiennent », ajoute Turner. « Et je pense que cette étude montre exactement pourquoi, en termes de DDX3X. »
Parce que les mutations du gène affectent largement les filles, dit Turner, une meilleure compréhension de sa fonction pourrait également aider à mettre en lumière les différences entre les sexes dans les conditions de développement du cerveau.
Références :
(1) Lennox A.L. et al. Neuron Epub avant impression (2020) PubMed
(2) Iossifov I. et al. Nature 515, 216-221 (2014) PubMed
(3) Snijders Blok L. et al. Am. J. Hum. Genet. 97, 343-352 (2015) PubMed








